Partial Agonists
In the classical receptor theory developed by Clark, it was assumed
that the effect of a drug is proportional to the fraction of receptors occupied by the drug and that maximal effect results
when all receptors are occupied. This assumption may only be true for a limited number of cases. To account for the discrepancies,
Ariëns introduced the term intrinsic activity to describe the relationship between the effect elicited by a drug and the concentration
of drug-receptor complexes.
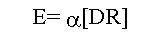
Where E is the effect; a is a proportionality constant or
the intrinsic activity of the drug; and [DR] the concentration of drug-receptor complex. This relationship addressed the observation
that some drugs do not elicit a maximal response even at apparently maximal receptor occupancies. These drugs are called partial
agonists.
What does a partial agonist look like?
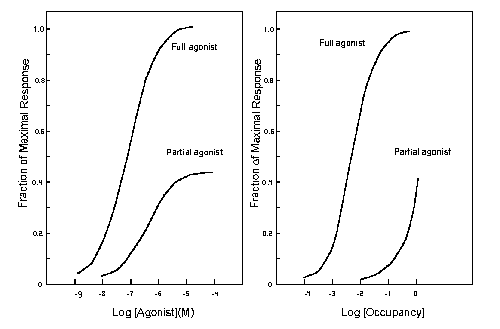
The figure below on the left shows the concentration effect curve
for a full agonist and a partial agonist, the binding affinity of the receptor for both drugs is the same. Notice the shift
to the right of the concentration effect curve for the partial agonist relative to the full agonist. On the right are plotted
the occupancy response curves for the data on the left. Note that the partial agonist in spite of achieving maximal occupancy,
elicit only 45 % of the maximal response.
Stephenson introduced the concept of efficacy as a property of the
drug to explain the nonlinear relationships between occupancy and response, and for the ability of a group of drugs (partial
agonists) to produce equal responses while occupying different proportions of the receptor population. According to Stephenson,
the response, R, of a tissue is a function of the stimulus, S, given to the tissue: R = f (S). S was
defined as the product of the efficacy of an agonist, e , multiplied by the fraction of receptors occupied by the agonist.
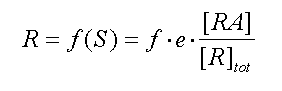
The differences in the effects of a drug in different tissues are
a reflection of the contribution of properties of the drug, properties of the receptor, and properties of a tissue in terms
of receptor density, and the coupling of receptor occupancy to the ultimate response.
Partial agonists by virtue of the occupation of a large number of
receptors without eliciting a response, competitively block the effects of agonists of higher intrinsic efficacies or full
agonists. Since both effects of the partial agonist are the result of the interaction with the same site, antagonism from
partial agonists should be observed at the same concentrations that produce the agonist effect. Therefore, the agonistic response
to a drug that occurs at concentrations different than those that produce antagonism, cannot be ascribed to interaction of
the drug with a single receptor.
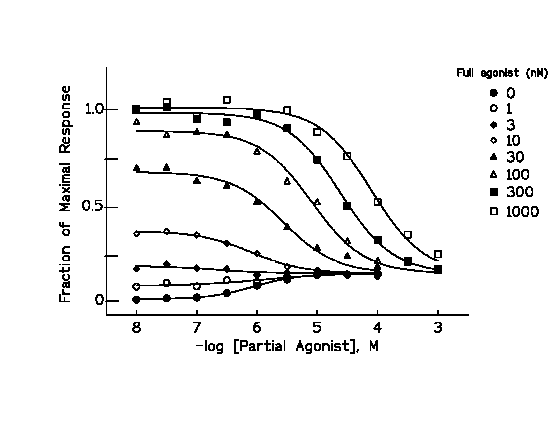
The figure above shows a series of concentration-effect curves for
a partial agonist in the absence and in the presence of different concentrations of a full agonist. The partial agonist produced
20 % of the maximal response. Note that the effects of the partial agonist in the presence of low concentrations of full agonist
(producing a response lower than the maximal response of the partial agonist i.e. 20 %) are additive to those of the full
agonist up to the maximal response of the partial agonist (20%). In contrast, the partial agonist in the presence of concentration
of the full agonist that produce responses higher than those of the partial agonist (more than 20%) has antagonistic effects,
decreasing the response of the full agonist to the level of the maximal response of the partial agonist (20%). Note also that
the agonistic and antagonistic effects of the partial agonist are produced at the same concentration range at low concentrations
of the full agonist. The increase in concentrations of the partial agonist required to antagonize the effects of increasingly
higher concentrations of full agonist are indicative of the competitive nature of the effect.
Estimating the potency of partial agonists
Several approaches have been used to estimate the potency (equilibrium
dissociation constants) of partial agonists (see Chapter 2 in Lee Limbird’s book).
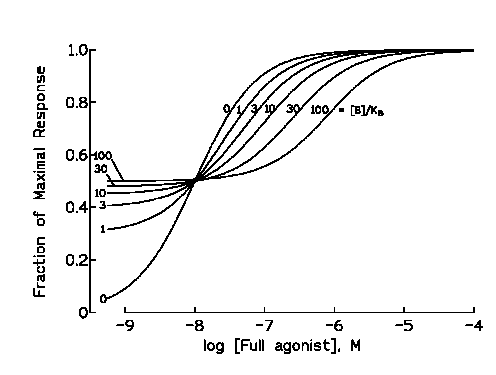
The figure above shows the effects of various concentrations of
a partial agonist on the concentration response curves for a full agonist. Note the dual effects of the partial agonist in
this plot. The increase in the response produced by the partial agonist in the absence of the full agonist, and the shifts
to the right in the concentration effect curves for the full agonist which can provide dose-ratios for Schild analysis as
indicated in the previous section.
This approach nonetheless has some caveats, first, which region
of the concentration effect curve of the full agonist should be used to measure the dose ratios? It has been observed that
the dose ratio taken from the half-maximal concentration of the full agonist dose response curves produce the most unambiguous
approach; second, the intrinsic efficacy of the partial agonist could introduce an error in the estimation of the KB. This
error is proportional to the magnitude of the intrinsic efficacy of the partial agonist and to the efficiency of the coupling
of the tissue stimulus-response mechanism. In general, for low efficacy partial agonist this method produces accurate estimations
of the KB. A most unambiguous method to determine the KB for a partial agonist consist in to irreversibly block or inactivate
a sufficient fraction of the receptor population so that the partial agonist no longer produces a response but that still
permit to observe the effects of a full agonist. The interaction of the partial agonist with its receptor can then be studied
as a competitive antagonist of the full agonist as indicated in the previous section.
Decreasing receptor concentration
Several approaches have been used successfully to decrease the number
of the receptor population. The primary criterion for the validity of any of these approaches is that the treatment will eliminate
a fraction of the receptor population, yet should not affect coupling of the remaining receptors to any of the response system
(e.g., an equal number of agonist-receptor complexes elicit equal responses before and after the irreversible receptor blockade).
Some possible approaches include:
- Receptor-selective irreversible antagonists (unfortunately
such drugs may not be available for most receptor subtypes).
- Non-selective irreversible receptor antagonists.
(Question: how can you use what you have learned about receptor binding to minimize the artifacts induced by non-selective
irreversible antagonists?)
- Non-ligand inactivators that cross-linking the ligand
to the receptor. For example, a bifunctional reagent, such as succinimidyl suberimidate may achieve a specific irreversible
blockade.
- Non-selective biochemical inactivation. Reagents such
as N-ethylmaleimide or other residue-directed reagents, or limited proteolysis, have sometimes been used.
- Immunotitration. An excellent method if a selective
deactivating antibody is available.
- Molecular control. With a cloned, expressed receptor,
one can often manipulate (or at least select) for varying levels of receptor expression.
The primary criterion for the validity of any of these approaches
is that the treatment will only eliminate a fraction of the receptor population and must not affect in any way the receptor
coupling of the remaining receptors, or the response system, so that equal number of agonist-receptor complexes elicit equal
responses before and after the irreversible receptor blockade.


