


Aromaticity |
|---|
The adjective "aromatic" is used by organic chemists in a rather different way than it is normally applied. It has its origin in the observation that certain natural substances, such as cinnamon bark, wintergreen leaves, vanilla beans and anise seeds, contained fragrant compounds having common but unexpected properties. Cinnamon bark, for example, yielded a pleasant smelling compound, formula C9H8O, named cinnamaldehyde. Because of the low hydrogen to carbon ratio in this and other aromatic compounds (note that the H:C ratio in an alkane is >2), chemists expected their structural formulas would contain a large number of double or triple bonds. Since double bonds are easily cleaved by oxidative reagents such as potassium permanganate or ozone, and rapidly add bromine and chlorine, these reactions were applied to these aromatic compounds. Surprisingly, products that appeared to retain many of the double bonds were obtained, and these compounds exhibited a high degree of chemical stability compared with known alkenes and cycloalkenes (aliphatic compounds). On treatment with hot permanganate solution, cinnamaldehyde gave a stable, crystalline C7H6O2 compound, now called benzoic acid. The H:C ratio in benzoic acid is <1, again suggesting the presence of several double bonds. Benzoic acid was eventually converted to the stable hydrocarbon benzene, C6H6, which also proved unreacive to common double bond transformations, as shown below. For comparison, reactions of cyclohexene, a typical alkene, with these reagents are also shown (green box). As experimental evidence for a wide assortment of compounds was acquired, those incorporating this exceptionally stable six-carbon core came to be called "aromatic".

If benzene is forced to react by increasing the temperature and/or by addition of a catalyst, It undergoes substitution reactions rather than the addition reactions that are typical of alkenes. This further confirms the previous indication that the six-carbon benzene core is unusually stable to chemical modification. The conceptual contradiction presented by a high degree of unsaturation (low H:C ratio) and high chemical stability for benzene and related compounds remained an unsolved puzzle for many years. Eventually, the presently accepted structure of a regular-hexagonal, planar ring of carbons was adopted, and the exceptional thermodynamic and chemical stability of this system was attributed to resonance stabilization of a conjugated cyclic triene.
Benzene: |
 |
|---|
Here, two structurally and energetically equivalent electronic structures for a stable compound are written, but no single structure provides an accurate or even an adequate representation of the true molecule. The six-membered ring in benzene is a perfect hexagon (all carbon-carbon bonds have an identical length of 1.40 Ĺ). The cyclohexatriene contributors would be expected to show alternating bond lengths, the double bonds being shorter (1.34 Ĺ) than the single bonds (1.54 Ĺ). An alternative representation for benzene (circle within a hexagon) emphasizes the pi-electron delocalization in this molecule, and has the advantage of being a single diagram. In cases such as these, the electron delocalization described by resonance enhances the stability of the molecules, and compounds composed of such molecules often show exceptional stability and related properties.
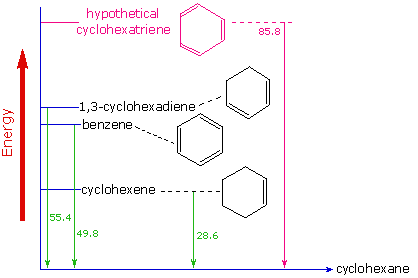
Evidence for the enhanced thermodynamic stability of benzene was obtained from measurements of the heat released when double bonds in a six-carbon ring are hydrogenated (hydrogen is added catalytically) to give cyclohexane as a common product. In the following diagram cyclohexane represents a low-energy reference point. Addition of hydrogen to cyclohexene produces cyclohexane and releases heat amounting to 28.6 kcal per mole. If we take this value to represent the energy cost of introducing one double bond into a six-carbon ring, we would expect a cyclohexadiene to release 57.2 kcal per mole on complete hydrogenation, and 1,3,5-cyclohexatriene to release 85.8 kcal per mole. These heats of hydrogenation would reflect the relative thermodynamic stability of the compounds. In practice, 1,3-cyclohexadiene is slightly more stable than expected, by about 2 kcal, presumably due to conjugation of the double bonds. Benzene, however, is an extraordinary 36 kcal/mole more stable than expected. This sort of stability enhancement is now accepted as a characteristic of all aromatic compounds.
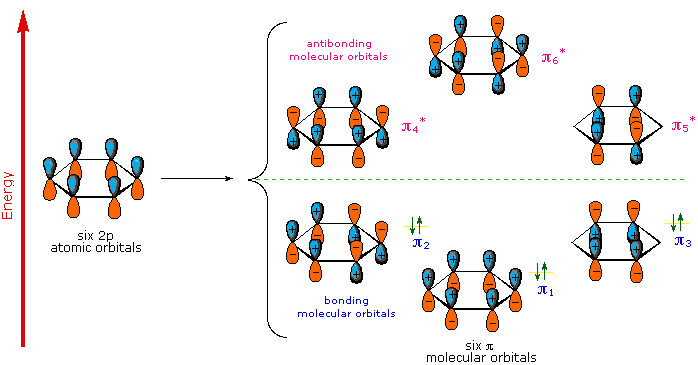
A molecular orbital description of benzene provides a more satisfying and more general treatment of "aromaticity". We know that benzene has a planar hexagonal structure in which all the carbon atoms are sp2 hybridized, and all the carbon-carbon bonds are equal in length. As shown below, the remaining cyclic array of six p-orbitals ( one on each carbon) overlap to generate six molecular orbitals, three bonding and three antibonding. The plus and minus signs shown in the diagram do not represent electrostatic charge, but refer to phase signs in the equations that describe these orbitals (in the diagram the phases are also color coded). When the phases correspond, the orbitals overlap to generate a common region of like phase, with those orbitals having the greatest overlap (e.g. π1) being lowest in energy. The remaining carbon valence electrons then occupy these molecular orbitals in pairs, resulting in a fully occupied (6 electrons) set of bonding molecular orbitals. It is this completely filled set of bonding orbitals, or closed shell, that gives the benzene ring its thermodynamic and chemical stability, just as a filled valence shell octet confers stability on the inert gases.
Benzene rings may be joined together (fused) to give larger polycyclic aromatic compounds. A few examples are drawn below, together with the approved numbering scheme for substituted derivatives. The peripheral carbon atoms (numbered in all but the last three examples) are all bonded to hydrogen atoms. The six benzene rings in coronene are fused in a planar ring; whereas the six rings in hexahelicene are not joined in a larger ring, but assume a helical turn, due to the crowding together of the terminal ring atoms. This helical configuration renders the hexihelicene molecule chiral, and it has been resolved into stable enantiomers having specific rotations of 3700ş.
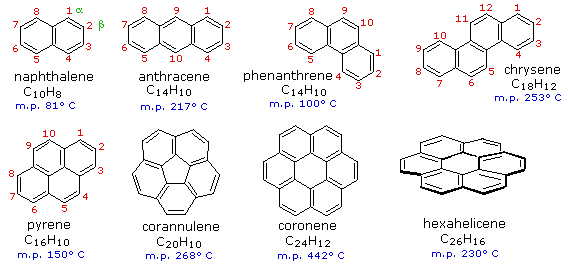
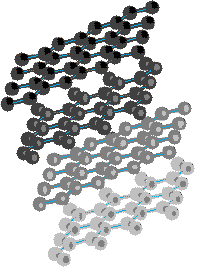
As these extended aromatic compounds become larger, the ratio of hydrogen to carbon decreases. For example, the symmetrical hexacyclic compound coronene has a H/C ratio =1/2, compared with 1 for benzene. If we were to imagine fused ring systems of this kind to be further extended in space, the H/C ratio would approach zero, and the resulting compound would be a form of carbon. Such a carbon allotrope exists and is called graphite. Another well-characterised carbon allotrope is diamond. The structures for these two forms of carbon are very different, and are displayed below. Diamond is an extended array of sp3 hybridized carbon atoms; whereas, graphite consists of overlapping sheets of sp2 hybridized carbon atoms arranged in a hexagonal pattern.
| Diamond | Graphite | |
|---|---|---|
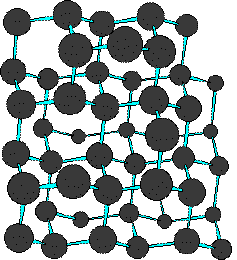 |
 |
A comparison of the coronene and corannulene models discloses an interesting difference in their shapes. Coronene is absolutely flat and, aside from the peripheral hydrogens, resembles a layer of graphite. Its very high melting point reflects this resemblance. Corannulene, on the other hand, is slightly curved, resulting in a bowl-like shape.
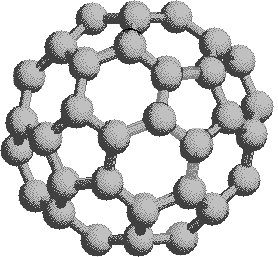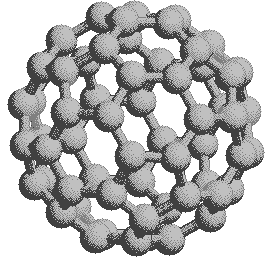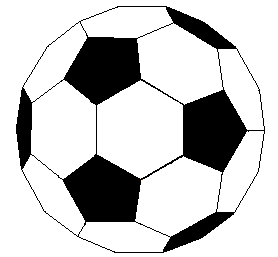 |
If we
extend the structure of corannulene by adding similar cycles of five
benzene rings, the curvature of the resulting molecule should
increase, and eventually close into a sphere of carbon atoms. The
archtypical compound of this kind (C60) has been named buckminsterfullerene
because of its resemblance to the geodesic structures created by
Buckminster Fuller. It is a member of a family of similar carbon
structures that are called fullerenes. These materials
represent a third class of carbon allotropes. Alternating views of
the C60 fullerene structure are shown
on the right, together with a soccer ball-like representation of the
12 five and 20 six-membered rings composing its surface. By clicking
on this graphic, a Chime model of C60
will be displayed.
Although C60 is
composed of fused benzene rings its chemical reactivity resembles
that of the cycloalkenes more than benzene. Indeed, exposure to
light and oxygen slowly degrade fullerenes to cage opened products.
Most of the reactions thus far reported for C60 involve addition to, rather than substitution
of, the core structure. These reactions include hydrogenation,
bromination and hydroxylation. Strain introduced by the curvature of
the surface may be responsible for the enhanced reactivity of
C60.
. Larger fullerenes, such as
C70, C76, C82 &
C84have elipsoidal or distorted
spherical structures, and fullerene-like assemblies up to
C240 have been detected. A
fascinating aspect of these structures is that the space within the
carbon cage may hold atoms, ions or small molecules. Such species
are called endohedral fullerenes. The cavity of C60 is relatively small, but encapsulated helium,
lithium and atomic nitrogen compounds have been observed. Larger
fullerenes are found to encapsulate lanthanide metal
atoms.
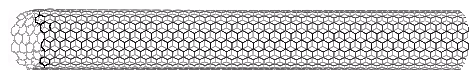
Interest in the fullerenes has led to the discovery of a related group of carbon structures referred to as nanotubes. As shown in the following illustration, nanotubes may be viewed as rolled up segments of graphite. The chief structural components are six-membered rings, but changes in tube diameter, branching into side tubes and the capping of tube ends is accomplished by fusion with five and seven-membered rings. Many interesting applications of these unusual structures have been proposed.
Many unsaturated cyclic compounds have exceptional properties that we now consider characteristic of "aromatic" systems. The following cases are illustrative:
Compound |
Structural Formula |
Reaction with Br2 |
Thermodynamic Stabilization | |||
|---|---|---|---|---|---|---|
| 1,3-Cyclopentadiene | 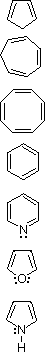 |
Addition ( 0 şC ) | Slight | |||
| 1,3,5-Cycloheptatriene | Addition ( 0 şC ) | Slight | ||||
| 1,3,5,7-Cyclooctatetraene | Addition ( 0 şC ) | Slight | ||||
| Benzene | Substitution | Large | ||||
| Pyridine | Substitution | Large | ||||
| Furan | Substitution ( 0 şC ) | Moderate | ||||
| Pyrrole | Substitution | Moderate |
The first three compounds (cyclic polyenes) have properties associated with alkenes in general. Each reacts readily with bromine to give addition products, as do most alkenes. The thermodynamic change on introducing double bonds into the carbon atom ring is also typical of alkenes (a destabilization of ca. 26 kcal/mol for each double bond). Conjugation offsets this increase in energy by a small amount (4-6 kcal/mol).
The remaining four compounds exhibit very different properties, and are considered aromatic. Benzene and pyridine are relatively unreactive with bromine, requiring heat and/or catalysts to force reaction, the result of which is substitution rather than addition. Furan and pyrrole react more rapidly with bromine, but they also give substitution products. This tendency to favor substitution rather than addition suggests that the parent unsaturated ring system has exceptional stability. Thermodynamic measurements support this conclusion. The enhanced stability, often referred to as aromatic stabilization, ranges (in the above cases) from a low of 16 kcal/mol for furan to 36 kcal/mol for benzene.
A planar (or near planar) cycle of sp2 hybridized atoms, the p-orbitals of which are oriented parallel to each other. These overlapping p-orbitals generate an array of π-molecular orbitals.
These π-orbitals are occupied by 4n+2 electrons (where n is an integer or zero). This requirement is known as The Hückel Rule. All the aromatic compounds discussed above have 6 π-electrons (n=1).
1,3-Cyclopentadiene and 1,3,5-cycloheptatriene both fail to meet the first requirement, since one carbon atom of each ring is sp3 hybridized and has no p-orbital. Cyclooctatetraene fails both requirements, although it has a ring of sp2 hybridized atoms. This molecule is not planar ( a geometry that would have 135ş bond angles ). Angle strain is relieved by adopting a tub-shaped conformation; consequently, the p-orbitals can only overlap as isolated pairs, not over the entire ring. Furthermore, cyclooctatetraene has 8 π-electrons, a number not consistent with the Hückel Rule.
Benzene is the archetypical aromatic compound. It is planar, bond angles=120ş, all carbon atoms in the ring are sp2 hybridized, and the pi-orbitals are occupied by 6 electrons. Pyridine is similar to benzene, but it is essential to recognize that the non-bonding electron pair on nitrogen occupies a sp2 orbital lying in the plane of the ring, and is not part of the π-electron system. Thus pyridine has 6 π-electrons and 2 non-bonding sp2 electrons.
Furan and pyrrole have five-membered rings, in which one atom is not carbon (a heteroatom) and has at least one pair of non-bonding valence shell electrons. By hybridizing this heteroatom to a sp2 state, a p-orbital occupied by a pair of electrons and oriented parallel to the carbon p-orbitals is created. The resulting planar ring meets the first requirement for aromaticity, and the π-system is occupied by 6 electrons, 4 from the two double bonds and 2 from the heteroatom, thus satisfying the Hückel Rule.
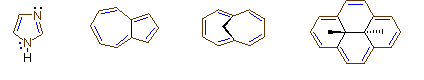
Four other examples of aromatic compounds are shown above. The sp2 hybridized ring atoms are connected by brown bonds, the π-electron pairs and bonds that constitute the aromatic ring are colored blue. Bonds and electron pairs that are not part of the aromatic annulene are black. The first example is imidazole, a heterocycle having two nitrogen atoms. Note that only one of the nitrogen non-bonding electron pairs is used for the aromatic π-electron sextet. The other electron pair (colored black) behaves similarly to the electron pair in pyridine. The second and third compounds have ten-membered rings, which are forced to be planar, or nearly planar, by the bridging bond or methylene group. These are 10 π-electron systems that fit the aromaticity requirements. Finally, the last example has a fourteen-membered ring with 7 double bonds. The bridging atoms and bonds force it to be planar. The number of π-electrons in this aromatic system is 14.
Monocyclic compounds made up of alternating conjugated double bonds are called annulenes. Benzene and 1,3,5,7-cyclooctatetraene are examples of annulenes; they are named [6]annulene and [8]annulene respectively, according to a general nomenclature system in which the number of pi-electrons in an annulene is designated by a number in brackets. Some annulenes are aromatic (e.g. benzene), but many are not due to non-planarity or a failure to satisfy the Hückel Rule. Non-planarity of a simple ring may be corrected by bridging bonds or atoms, as noted above.
Carbanions and carbocations may also show aromatic stabilization. Some examples are:
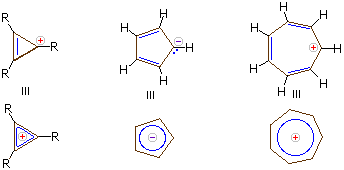
The three-membered ring cation has 2 π-electrons and is surprisingly stable, considering its ring strain. Cyclopentadiene is as acidic as ethanol, reflecting the stability of its 6 π-electron conjugate base. Salts of cycloheptatrienyl cation (tropylium ion) are stable in water solution, again reflecting the stability of this 6 π-electron cation.
Conjugated ring systems having 4n π-electrons (e.g. 4, 8, 12 etc. electrons) not only fail to show any aromatic properties, but appear to be less stable and more reactive than expected. As noted above, 1,3,5,7-cyclooctatetraene is non-planar and adopts a tub-shaped conformation. The compound is readily prepared, and undergoes addition reactions typical of alkenes. Catalytic hydrogenation of this tetraene produces cyclooctane. Planar bridged annulenes having 4n π-electrons have proven to be relatively unstable. Examples of 8 and 12-π-electron systems are shown below, together with a similar 10 π-electron aromatic compound.
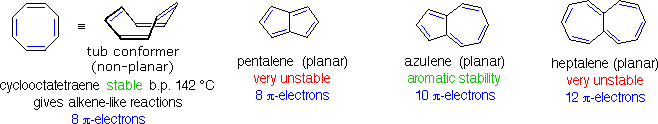
The simple
C8H6
hydrocarbon pentalene does not exist as a stable compound, and its
hexaphenyl derivative is air sensitive. The 12-π-electron analog
heptalene has been prepared, but is also extremely reactive (more so
than cyclooctatetraene). On the other hand, azulene is a stable
10-π-electron hydrocarbon that incorporates structural features of
both pentalene and heptalene. Azuleneis a stable blue crystalline
solid that undergoes a number of typical aromatic substitution
reactions. The unexpected instability of 4n π-electron annulenes has
been termed "antiaromaticity".
Other examples may be
cited. Thus, all attempts to isolate 1,3-cyclobutadiene have yielded
its dimer, or products from reactions with other compounds
introduced into the reaction system. Similarly, cyclopentadienyl
cation (4 π-electrons) and cycloheptatrienyl anion (8 π-electrons)
show very high reactivity when forced to form.
Nucleophilicity & Basicity |
|---|
Recall the definitions of electrophile and nucleophile:
|
Electrophile:
An electron deficient atom, ion or molecule that has an
affinity for an electron pair, and will bond to a base or
nucleophile. |
Many functional groups have weakly electrophilic carbon atoms (colored red in the following examples). These include alkyl halides and sulfonate esters {C-X and C-OSO2R}, as well as carbonyl compounds such as aldehydes and ketones {C=O}. These electrophilic functions may react with nucleophiles (bases) in two distinct ways:
|
(i) Substitution or
addition at carbon (this reflects nucleophilicity)
|
Because
these electrophilic reactants are weak, such reactions normally
require strong nucleophiles or bases to proceed. However, if a
preliminary ionization to a strongly electrophilic carbocation
occurs: [ C-X ——> C(+) + X(–)
]
or if the carbonyl group is converted to its more electrophilic
conjugate acid: [ C=O + A(+)
——> (+)C-O-A]
then reactions with much weaker
nucleophiles or bases may take place.
Some
confusion in distinguishing basicity (base strength) and
nucleophilicity (nucleophile strength) is inevitable. Since basicity
is a less troublesome concept; it is convenient to start with it.
Basicity refers to the ability of a base to accept a
proton. Basicity may be related to the pKa of the corresponding conjugate acid, as shown
below. The strongest bases have the weakest conjugate acids and
vice versa. The range of basicities included in the following
table is remarkable, covering over fifty powers of ten!
In an
acid-base equilibrium the weakest acid and the weakest base will
predominate (they will necessarily be on the same side of the
equilibrium). Learning the pKa values
for common compounds provides a useful base on which to build an
understanding of acid-base factors in reaction mechanisms.
| Base | I (–) | Cl (–) | H2O | CH3CO2(–) | RS(–) | CN(–) | RO(–) | NH2(–) | CH3(–) |
|---|---|---|---|---|---|---|---|---|---|
| Conj. Acid | HI | HCl | H3O(+) | CH3CO2H | RSH | HCN | ROH | NH3 | CH4 |
| pKa | -9 | -7 | -1.7 | 4.8 | 8 | 9.1 | 16 | 33 | 48 |

Nucleophilicity is a more complex property. It commonly refers to the rate of substitution reactions at the halogen-bearing carbon atom of a reference alkyl halide, such as CH3-Br. Thus the nucleophilicity of the Nu:(–) reactant in the following substitution reaction varies as shown in the chart below:
 |
|---|
|
Nucleophilicity: CH3CO2 (–) < Cl(–) < Br(–) < N3(–) < CH3O(–) < CN(–) < I(–) < CH3S(–) |
 |
The reactivity range encompassed by these reagents is over 5,000 fold, thiolate being the most reactive. Clearly, there are significant differences between these nucleophilicities and the basicities discussed above.
|
Some useful trends have
been documented: |
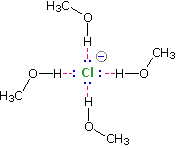 Solvation of nucleophilic anions markedly influences their
reactivity. The nucleophilicities cited above were obtained from
reactions in methanol solution. Polar, protic solvents such as water
and alcohols solvate anions by hydrogen bonding interactions, as
shown in the diagram on the right. These solvated species are more
stable and less reactive than the unsolvated "naked" anions. Polar,
aprotic solvents such as DMSO (dimethyl sulfoxide), DMF
(dimethylformamide) and acetonitrile do not solvate anions nearly as
well as methanol, but provide good solvation of the accompanying
cations. Consequently, most of the nucleophiles discussed here react
more rapidly in solutions prepared from these solvents. These
solvent effects are more pronounced for small basic anions than for
large weakly basic anions. Thus, for reaction in DMSO solution we
observe the following reactivity order:
Solvation of nucleophilic anions markedly influences their
reactivity. The nucleophilicities cited above were obtained from
reactions in methanol solution. Polar, protic solvents such as water
and alcohols solvate anions by hydrogen bonding interactions, as
shown in the diagram on the right. These solvated species are more
stable and less reactive than the unsolvated "naked" anions. Polar,
aprotic solvents such as DMSO (dimethyl sulfoxide), DMF
(dimethylformamide) and acetonitrile do not solvate anions nearly as
well as methanol, but provide good solvation of the accompanying
cations. Consequently, most of the nucleophiles discussed here react
more rapidly in solutions prepared from these solvents. These
solvent effects are more pronounced for small basic anions than for
large weakly basic anions. Thus, for reaction in DMSO solution we
observe the following reactivity order:
|
Nucleophilicity: I(–) < Br(–) < Cl(–) ~ N3(–) < CH3CO2 (–) < CN(–) ~ CH3S(–) < CH3O(–) |
|
|---|
Note that this order is roughly the order of increasing basicity (see above).
Acid-Base Catalysis |
|---|
As we have noted, many common organic reactions proceed by bonding between nucleophilic and electrophilic sites in the reactant molecules. Three examples are shown in equations 1 through 3; electrophiles are colored red, and nucleophiles are colored blue.
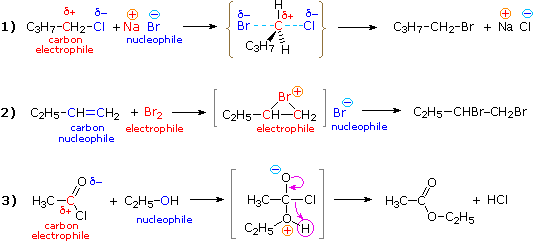
Reaction #1 is an example of a SN2 substitution reaction. The electrophilic carbon of 1-chlorobutane is attacked by the nucleophilic bromide anion in a single-step displacement process. The curly brackets enclose a structure for the transition state in this reaction.
Reactions #2 and #3 are two-step sequences. In the former addition reaction, bromine (an electrophile) attacks the nucleophilic double bond of 1-butene to give an electrophilic cyclic-bromonium intermediate (enclosed in square brackets) accompanied by a nucleophilic bromide ion. This ion-pair is very short-lived, another nucleophile-electrophile bonding reaction leads to the product (1,2-dibromobutane). In reaction #3 (a substitution reaction) the electrophilic carbonyl carbon atom bonds to the nucleophilic oxygen atom of ethyl alcohol to give an intermediate (in square brackets) that eliminates HCl, yielding the ester ethyl acetate. In all of these examples the reactivity of the electrophiles and nucleophiles is sufficient to allow reaction to proceed spontaneously at room temperature.
It is not difficult, however, to find very similar combinations of compounds that either fail to react at all, or do so extremely slowly. Equations 4 through 6 illustrate this behavior for analogs of the first three reactions.
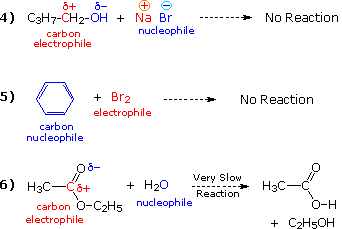
The factors that influence such differences in reactivity may be complex, but in the above cases are largely due to a poor anionic leaving group (eq. 4); aromatic stabilization and reduced nucleophilicity of a conjugated π-electron system (eq. 5); and reduced electrophilic character of a substituted carbonyl group (eq. 6).
First,
compare reaction #4 with #1. Since oxygen is slightly more
electronegative than chlorine (3.5 vs. 2.8 on the Pauling scale), we
might expect a C-O bond to be more polar than a C-Cl bond. A better
measure of the electrophilic character of a carbon atom in a
compound comes from nmr chemical shifts (both 1H & 13C),
and these indicate that oxygen and chlorine substituents exert
similar effects when bonded to sp3
hybridized carbon atoms. In any event, the failure of
reaction #4 cannot be due to differences in the electrophilicity and
nucleophilicity of the reactants.
The key factor here is the
stability of the leaving anion (chloride vs. hydroxide). We know
that HCl is a much stronger acid than water (by more than 15 powers
of ten), and this difference will be reflected in reactions that
generate their conjugate bases. Thus, chloride anion is much more
stable and less reactive than is hydroxide anion, so the former is a
better and more common leaving group. Indeed, the failure of
alcohols to undergo SN2 substitution
reactions makes them useful solvents for many such reactions,
including #1.
In the case of reaction #5, the aromatic stabilization of the benzene ring makes it less susceptible to attack by electrophiles than are simple alkenes. Thus, elemental bromine is not sufficiently electrophilic to induce a reaction with benzene, even though the latter is nucleophilic.
Lastly,
reactions #3 and #6 illustrate differences in the reactivity of
carbonyl compounds. We know that the carbon atom of a carbonyl group
is electrophilic and undergoes reaction with a variety of
nucleophiles. However, this electrophilic character may be enhanced
or diminished by substituents. If we take saturated aldehydes
(RCH=O) as a reference, the additional alkyl substituent present in
ketones slightly reduces this electrophilicity, but the general
reactivity pattern of these classes is similar. On the other hand, a
chlorine substituent is inductively electron withdrawing and
increases the electrophilicity of the carbonyl carbon significantly.
Thus, acid chlorides are very reactive with a wide range of
nucleophiles, including water and alcohols (eq. 3).
The oxygen
and nitrogen substituents present in esters and amides have a
similar inductive effect, but also a pronounced electron donating
character through a resonance interaction. Consequently, the
carbonyl carbon atom becomes less electrophilic, and these
functional groups are less reactive than other carbonyl compounds.
Thus, the failure of ethyl acetate to react with water (eq. 6)
reflects the reduced electrophilic character of its carbonyl
group.
Fortunately, these retarding factors can often be overcome by acid or base catalysis, which in general enhances electrophilicity (acids) or nucleophilicity (bases). Equations 7 through 9 show how this tactic may be effectively applied to the unreactive examples given above.
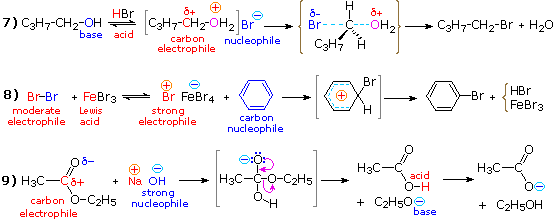
In strong
acid, reaction #4 proceeds nicely, as shown in equation 7. This is
because the leaving group has changed from hydroxide anion to water
(the acidity of the conjugate acid H3O(+) is nearly
that of HCl).
In the second
example, there are two obvious ways of circumventing the failure of
benzene to react with bromine:
• The bromine can be made more electrophilic
• The benzene ring can be made more
nucleophilic.
The first tactic can be implemented by mixing
bromine with ferric bromide, a Lewis acid catalyst. This generates
the bromonium cation, Br(+), a
powerful electrophile. Equation #8 illustrates this approach.
The
second tactic requires that the benzene ring be activated (made more
nucleophilic) by substitution with an electron donating group such
as OH or NH2. For example, we find
that phenol (hydroxybenzene) reacts rapidly with bromine in the
absence of any catalyst.
Finally,
we can see that there are two ways of facilitating the ester
hydrolysis reaction.:
•We can use a stronger nucleophile than water, such as
hydroxide anion.
•We
can increase the electrophile reactivity by converting the ester to
its conjugate acid, CH3C(OH)OC2H5 (+).
Equation #9 shows the former approach,
which is an example of base catalysis. Acid catalysis of the
reaction also works well.