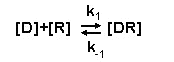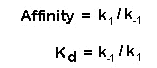RECEPTOR:
Any cellular macromolecule that a drug binds to initiate
its effects.
DRUG:
A chemical substance that interacts with a biological
system to produce a physiologic effect.
All drugs are chemicals but not all chemicals are drugs.
The ability to bind to a receptor is mediated by the chemical structure of the drug that allows it to interact with complementary
surfaces on the receptor. Drugs that interact with receptors can be classified as being either agonists or antagonists. Once
bound to the receptor an agonist activates or enhances cellular activity. Examples of agonist action are drugs that bind to
beta receptors in the heart and increase the force of myocardial contraction or drugs that bind to alpha receptors on blood
vessels to increase blood pressure. The binding of the agonist often triggers a series of biochemical events that ultimately
leads to the alteration in function. The biochemicals that initiate these changes are referred to as second messengers. Antagonists
have the ability to bind to the receptor but do not initiate a change in cellular function. Because they occupy the receptor,
they can prevent the binding and the action of agonists. Hence the term antagonist. Antagonists are also referred to as blockers.
 |
|
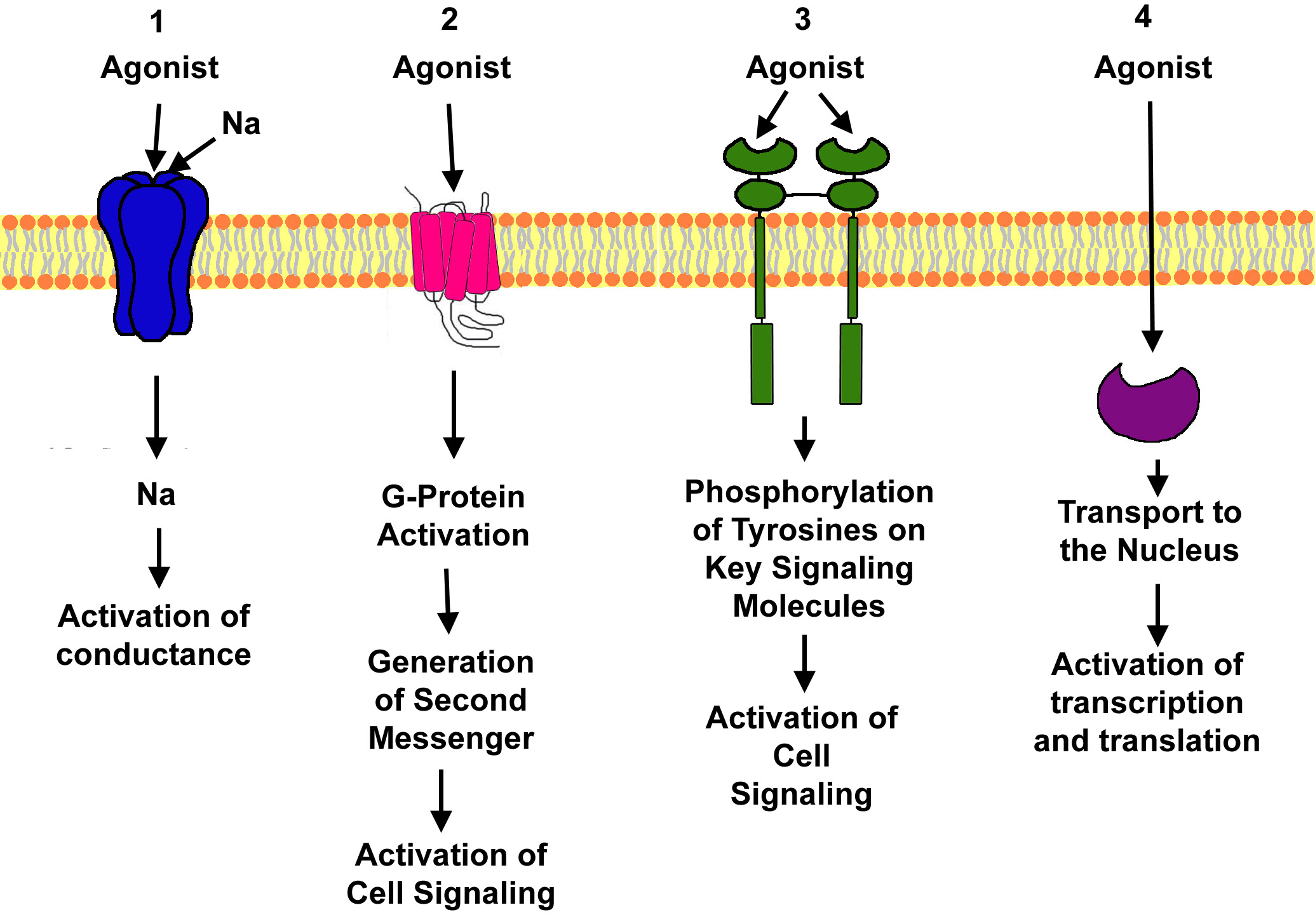
|
Factors Governing Drug Action
Two factors that determine the effect of a drug on physiologic processes
are affinity and intrinsic activity.Affinity is a measure of the tightness that a drug binds to the receptor.
Intrinsic
activity is a measure of the ability of a drug once bound to the receptor to generate an effect activating stimulus and producing
a change in cellular activity.
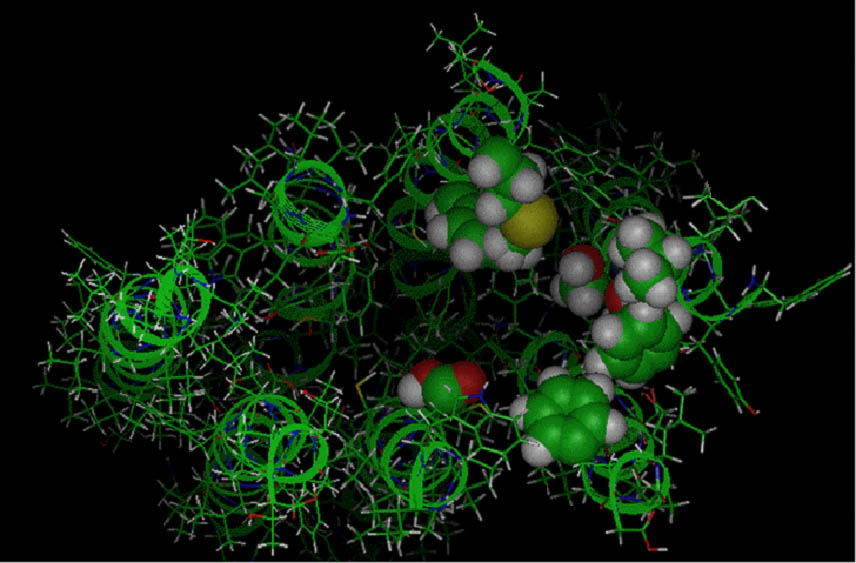 Affinity and intrinsic activity are independent properties of drugs. Agonists have both affinity, that is, the ability
to bind to the receptor, as well as intrinsic activity, the ability to produce a measurable effect. Antagonists, on the other
hand, only have affinity for the receptor. This property allows antagonists to bind to the receptor. However, because
antagonists do not have intrinsic activity at the receptor no effect is produced. Because they are bound to the receptor,
they can prevent binding of agonists. This is a diagram of a G-protein coupled receptor. Notice how the amino acids that make
up the receptor protein can contribute functional groups to allow a drug to bind to this receptor. Affinity and intrinsic activity are independent properties of drugs. Agonists have both affinity, that is, the ability
to bind to the receptor, as well as intrinsic activity, the ability to produce a measurable effect. Antagonists, on the other
hand, only have affinity for the receptor. This property allows antagonists to bind to the receptor. However, because
antagonists do not have intrinsic activity at the receptor no effect is produced. Because they are bound to the receptor,
they can prevent binding of agonists. This is a diagram of a G-protein coupled receptor. Notice how the amino acids that make
up the receptor protein can contribute functional groups to allow a drug to bind to this receptor. |
The binding of a drug to a receptor is determined by the following
forces:
- Hydrogen bonds
- Ionic bonds
- Van der Waals forces
- Covalent bonds
To bind to a receptor the functional group on a drug must interact
with complementary surfaces on the receptor. The binding of a drug, illustrated here as D, to the receptor, illustrated as
R, can be described by this expression.
| This is a reversible reaction and when at equilibrium,
the rate of drug-receptor complex formation [DR] is equal to the rate of drug-receptor complex dissociation. The rate of formation
of the drug-receptor complex is described by k1. The rate at which the drug receptor complex dissociates is described
by k-1. The binding of many, but not all, drugs to the receptor is a reversible process which reaches an equilibrium.
The practical consequence of this is when binding is in equilibrium the amount of drug bound to the receptor is constant.
Affinity is equal to the ratio of k1 and k-1.
Kd is the equilibrium dissociation constant and is the reciprocal of the affinity. It is an important term in
pharmacology. It is the term which can be used to describe the affinity of drugs to receptors. The units of the dissociation
constant are some measure of concentration such as molar, millimolar, micromolar, nanomolar and so forth. Dissociation
constants are usually small numbers, significantly less than 1, such as 1 x 10-8M or 10 nanomolar. There is an
inverse relationship between the Kd and affinity. The smaller the Kd, the greater the affinity. A drug that has a dissociation
constant of 1 nanomolar is said to have higher affinity than a drug that has a dissociation constant of 1 micromolar. This
is because 1 nanomolar is much smaller than 1 micromolar. |
By appropriate manipulation we can write the equation:
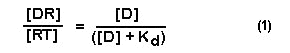
Where [D] is the concentration of drug, [DR] is the concentration
of receptor bound with drug, [RT] is the total number of receptors and Kd is the equilibrium dissociation constant. This equation
describes the binding of drugs to pharmacologic receptors and states that the amount of drug bound to the receptor is dependent
on the DRUG CONCENTRATION AND KD. When a drug is given at a concentration equal to its Kd, 50 % of the total receptor
population will be occupied by the drug.
We can make the assumption that the physiologic effect of the drug,
E, is proportional to the amount of drug bound to the receptor:
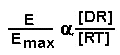
where Emax is the maximal obtainable effect when all receptors are
occupied. We can now write:
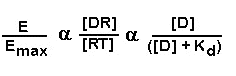
This states that the effect observed, E/Emax, is determined by the
concentration of the drug and the affinity (Kd) the drug possesses for the receptor. We will add one additional term to this
equation. That is the intrinsic activity term. Let us use the symbol e for intrinsic activity. Intrinsic activity therefore
will also modulate the drug effect. Intrinsic activity can be thought of as the strength that a drug can impart an activating
stimulus to a receptor.
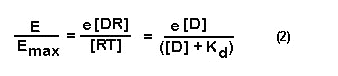
When a drug is given at a concentration equal to its Kd, 50 % of
the total receptor population will be occupied by the drug. At this concentration of agonist the value of E/Emax will be equal
to 50 % of the maximal response. This concentration is also referred to as the EFFECTIVE DOSE-50 OR ED50.
Agonists can be further divided into full and partial agonists:
Full Agonists: Compounds that are able to elicit the maximal response
of the tissue following receptor occupation and activation.
Partial Agonists: Compounds that produce an agonist action, but
are unable to elicit the full response of the tissue.
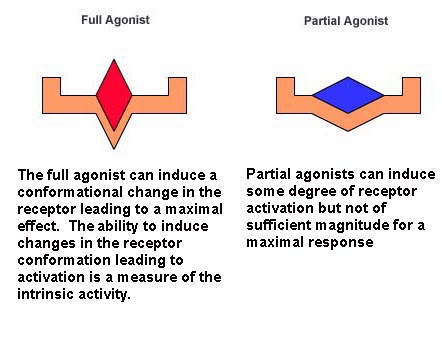
Drugs that are full agonists will have an intrinsic activity value
of 1. Partial agonists, that cannot produce the same maximal effect as full agonists will have intrinsic activity values less
than 1. Because partial agonists have e values less than 1, the value of E/Emax will be some fraction of the value obtained
with a full agonist.
Dose-Response Curves
Dose-response relationships are a common way to portray data in
both basic and clinical science. For example, a clinical study may examine the effect of increasing amounts of an analgesic
on pain threshold. To present the data, the concentration of the drug would be plotted on the x-axis and the effect on pain
threshold would be presented on the y-axis. A plot of drug concentration ([D]) versus effect (E/Emax) (or for that matter
DR/RT) is a rectangular hyperbola. Notice how the drug effect reaches a plateau or maximum. This is because there are a finite
number of receptors. Hence, the response must eventually reach a maximum. However, the hyperbolic plot is a cumbersome graph
because drug concentrations often vary over 100 to 1000-fold. This necessitates a long X-axis. To overcome this problem, the
log of the drug concentration is plotted versus the effect. A plot of the log of [D] versus E/Emax is a sigmoid curve.
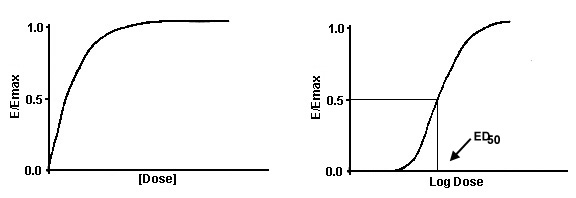
The position and shape of the curve are determined by the affinity
and the intrinsic activity. The concentration at which 50% of the maximal response occurred is referred to as the ED50 or
Effective Dose 50.
 |
The two
agonists NE and PE have different affinities but equal intrinsic activities. |
|
|
 |
Clonidine is a partial
agonist with higher affinity but lower intrinsic activity than methoxamine
|
Practical Implications of the Dose-Response Curve
Assume that a drug has an intrinsic activity of 1 and a Kd of 10
ng/ml. The drug is injected until it achieves a steady state level of 5 ng/ml. Assume also that the drug is in equilibrium
with the receptor compartment. At this blood level, calculated the number of receptors occupied and the effect that will be
observed.
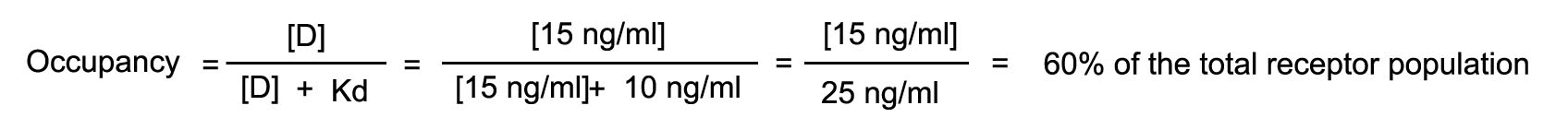
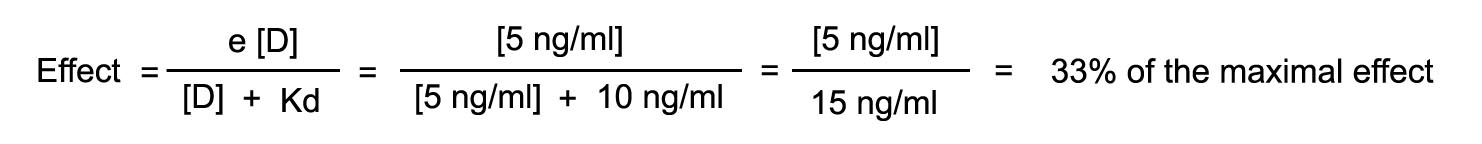
The rate of injection is increased such that the steady state blood
level is now 10 ng/ml. What will be the number of receptors occupied and observed effect?
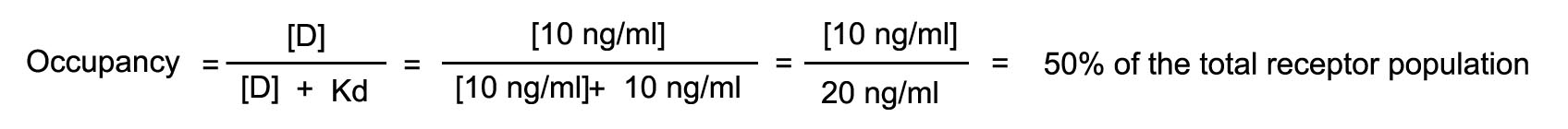
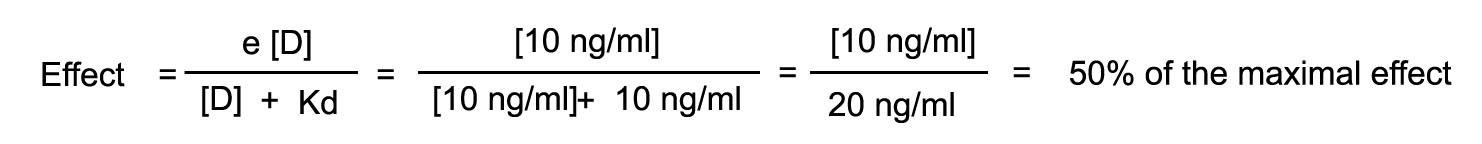
The rate of infusion is now increased to achieve a steady state
level fo 15 ng/ml. Calculate the number of receptors that will now be occupied as well as the effect.

Plot the log dose response curve.
Antagonists
 Antagonists exhibit affinity for the receptor but do not have intrinsic
activity at the receptor. An antagonist that binds to the receptor in a reversible mass-action manner is referred to as a
competitive antagonist. Because the antagonist does not have intrinsic activity, once it binds to the receptor, it blocks
binding of agonists to the receptor. A key point about competitive antagonists is that like agonists, they bind in a reversible
manner. This has important implications regarding the effect competitive antagonists have on the configuration of the dose-response
curve of agonists. Because competitive antagonists bind in a reversible manner, agonists, if given in high concentrations,
can displace the antagonist from the receptor and the agonist can then produce its effect. The antagonist action can, in effect,
be surmounted. Because the antagonist can be completely displaced, the agonist is still able to produce the same maximal effect
observed prior to antagonist treatment. However, because higher agonist concentrations were necessary to displace the antagonist,
the agonist dose-response curve is shifted to the right in the presence of a competitive antagonist. This can be illustrated
with two equilibrium equations:
Antagonists exhibit affinity for the receptor but do not have intrinsic
activity at the receptor. An antagonist that binds to the receptor in a reversible mass-action manner is referred to as a
competitive antagonist. Because the antagonist does not have intrinsic activity, once it binds to the receptor, it blocks
binding of agonists to the receptor. A key point about competitive antagonists is that like agonists, they bind in a reversible
manner. This has important implications regarding the effect competitive antagonists have on the configuration of the dose-response
curve of agonists. Because competitive antagonists bind in a reversible manner, agonists, if given in high concentrations,
can displace the antagonist from the receptor and the agonist can then produce its effect. The antagonist action can, in effect,
be surmounted. Because the antagonist can be completely displaced, the agonist is still able to produce the same maximal effect
observed prior to antagonist treatment. However, because higher agonist concentrations were necessary to displace the antagonist,
the agonist dose-response curve is shifted to the right in the presence of a competitive antagonist. This can be illustrated
with two equilibrium equations:
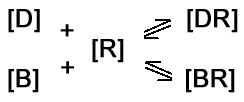
The antagonist [B] and agonist [D] are competing for the same limited
number of receptors [R]. The drug that binds to the receptor in the highest concentration will be determined by two factors.
These factors are the affinities of the agonist and antagonist for
the receptor and their relative concentrations. In the presence of a competitive antagonist equation #2 is modified as follows:
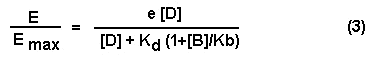
Where [B] is the concentration of antagonist and Kb is
the affinity exhibited by the antagonist for the receptor. Inspection of this equation will reveal that the affinity of the
agonist, Kd, is modified by the term (1+[B]/Kb). If the concentration of antagonist [B] is large in relation to its affinity
Kb, the term (1+[B]/Kb) will be large. Therefore, the major effect of an antagonist is to shift the dose-response curve for
an agonist to the right. The dose-response curve obtained in the presence of a competitive antagonist is parallel to the dose-response
curve obtained in the absence of antagonist. If the Kb is small and the concentration high, the antagonist will have a more
pronounced effect than if the Kb is large and the antagonist concentration is small. This also points out that large concentrations
of the agonist can overcome the actions of a competitive antagonist. Assume that the agonist, D, and the antagonist, B, have
equal affinity for the receptor. If the concentration of D is much larger than B, the value of E/Emax will not be significantly
decreased by the presence of the antagonist. This again illustrates that the actions of the competitive antagonist can be
surmounted by the agonist.
To summarize, the key features of a competitive antagonist are:
- Reversible binding to the receptor.
- The blockade can be overcome by increasing the agonist concentration.
- The maximal response of the agonist is not decreased.
- The agonist dose-response curve in the presence of a competitive
antagonist is displaced to the right parallel to the curve in the absence of agonist.
Irreversible Receptor Antagonists
Another type of antagonist is referred to as an irreversible
receptor antagonist. The properties of irreversible antagonists are markedly different from competitive antagonists. Irreversible
receptor antagonists are chemically reactive compounds. These ligands first bind to the receptor. Following this binding step,
the ligand then reacts with the functional groups of the receptor. The consequence of this chemical reaction is that the ligand
becomes covalently bound to the receptor. Because a chemical bond is formed, an irreversible ligand does not freely dissociate
from the receptor. It remains attached to the receptor for a long period of time. The synthesis of new receptor protein may
be required to generate a receptor free of an irreversible blocker. Because the ligand is covalently bound to the receptor,
the binding of agonists, and hence their pharmacologic activity, are blocked. Unlike competitive antagonists, the blocking
activity of irreversible receptor antagonists can not be overcome by increasing the agonist concentration. The antagonism
therefore cannot be overcome by increasing the agonist concentration. Recall, that the effect of an agonist is proportional
to the active drug-receptor complexes formed. Because an irreversible receptor antagonist reduces the total number of active
receptors, [RT], the maximal pharmacologic effect Emax is also decreased. The reduction in maximal agonist reponse is the
hallmark of irreversible antagonists. The shape of the dose-response curve is also altered because of this decrease in maximal
effect. The dose-response is shifted to the right and the maximal response is depressed.
To summarize, the properties of irreversible receptor blockers are:
- Chemically reactive compound, therefore covalently binds with the
receptor
- The receptor is irreversibly inactivated and the blockade can not
be overcome with increasing agonist concentration..
- Shifts the agonist dose-response curve to the right and depresses
maximal responsiveness.
Applications to Therapeutics
Few drugs interact with one and only one receptor. Such a drug would
be said to be specific, that is producing effects by specifically interacting with a single receptor. Most drugs interact
with several receptors and thus have the capability to produce distinctly different pharmacologic effects. Some of these effects
could be beneficial, some could be toxic. Such a drug would be said to be a selective. The factors that determine which
particular effect of a drug will be observed are the affinity and intrinsic activity of a drug .
To illustrate this point consider the following example. A drug
is capable of producing actions at 2 distinct receptors. At each of these receptors, the ligand has a different affinity as
well as pharmacologic effect.
Receptor System # 1:
KD = 40.0, intrinsic activity 1.0,effect- lowering of
systemic arterial blood pressure.
Receptor System # 2:
KD = 40.0, intrinsic activity 1.0,effect- lethal ventricular
arrhythmias.
Thus, this drug could either be a highly beneficial therapeutic
agent or a lethal poison. An overwhelming majority of drugs used in clinical practice produce their therapeutic effects due
to interactions at multiple pharmacologic receptors. This also illustrates
that whether the drug will be beneficial or poisonous depends on the skill and knowledge of the individual prescribing the
agent.
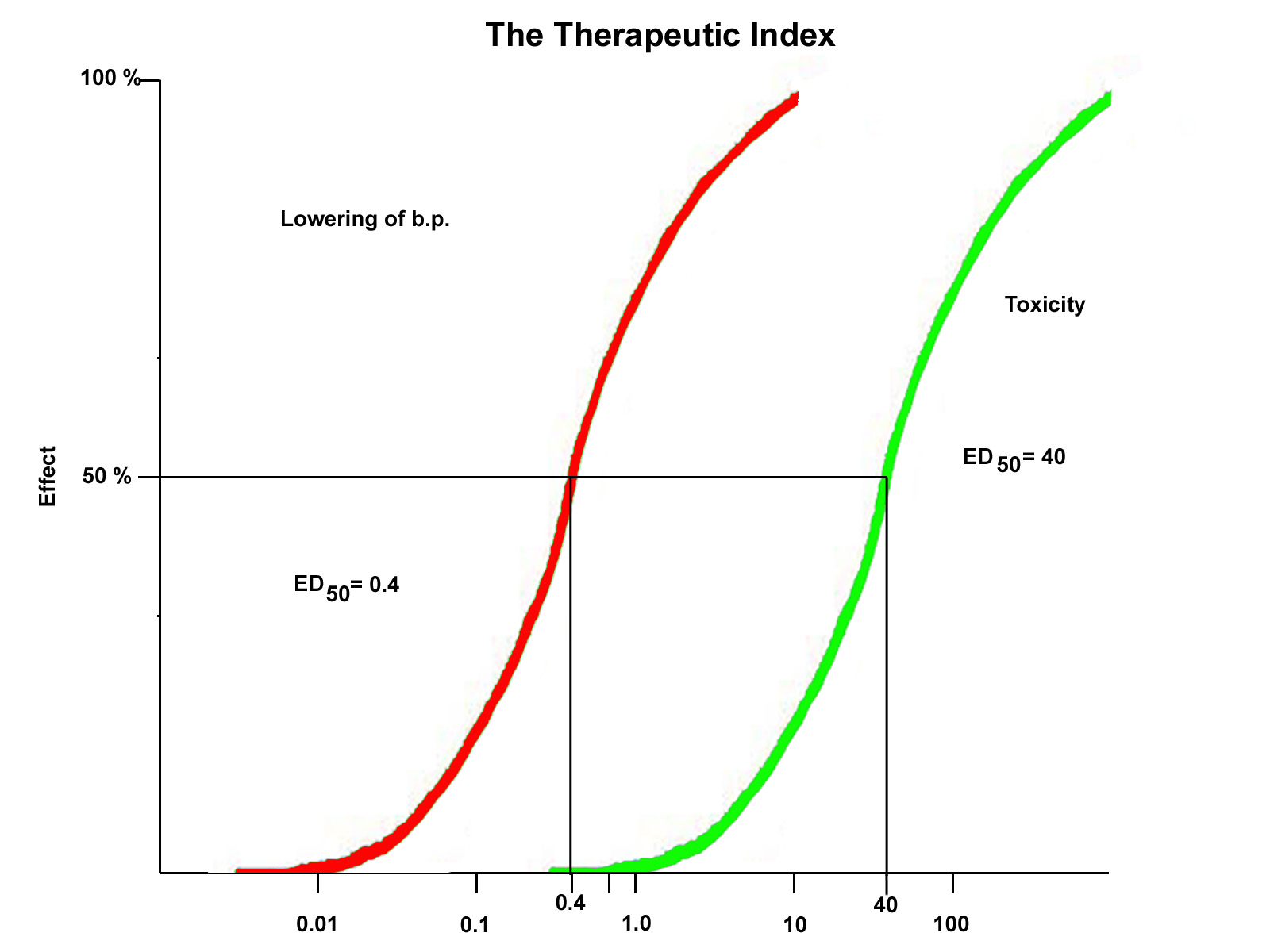
The Therapeutic Index
The therapeutic index is the ratio of the ED50 of a drug to produce
a toxic effect to the ED50 to produce a therapeutic effect. For the drug example above, the ED50 for the beneficial effect
of blood pressure lowering is 0.4 nM while the ED50 for toxicity is 40 nM. Therefore, the therapeutic index will be;
| TI = |
ED 50 (toxicity)
ED 50 (therapeutic) |
= |
40.0 nM
0.4 nM |
= 100 |